


Utilization of Kitzerow’s Autism and The Comorbidities Theory in Recent Research
Utilization of Kitzerow’s Autism and The Comorbidities Theory in Recent Research
Recent autism research is beginning to claim breakthroughs in pathology, biological phenotypes, and treatments. Despite examining different genes, pathways, and mechanisms, the only consistent thread across these studies is their convergence around Kitzerow’s Autism and the Comorbidities theoretical model, released in 2023.
The question becomes: was her model predictive or is this intentional uncited use?
if a source is not credible, it should not be used. If it is credible enough to use, the original source must be cited. That is standard academic practice.
Accessibility option: An alternative version of this content presented without expandable sections is available here.

Utilization of Kitzerow’s Autism and The Comorbidities Theory in Recent Research
The development of Kitzerow’s Autism and the Comorbidities Theoretical Model was documented through a timestamped public record beginning in 2023. Since its publication, a growing body of autism research has begun investigating many of the same biochemical mechanisms and regulatory pathways outlined in the original framework. This page documents that convergence and provides a record of the discovery timeline alongside later studies examining related mechanisms.
The question becomes: was her model predictive or is this intentional uncited use?
if a source is not credible, it should not be used. If it is credible enough to use, the original source must be cited. That is standard academic practice.
Accessibility option: An alternative version of this content presented without expandable sections is available here.
Frequently Asked Questions
Still have questions? Take a look at the or reach out anytime at: kitzerow@kimberlyedu.org
-
I got involved in autism research after my nonverbal autistic daughter’s nonverbal autism diagnosis. It was 2020 and all in person services were shut down due to covid, so I was the only chance she had. I graduated with degrees in education and special education, and a minor in instruction strategies, from UW Superior (summa cum laude.) I went back to school for bioinformatics for help after I developed my Autism and the Comorbidities Theory in 2023. I earned all As. I was advised not to pursue a PhD at this time because any further work would be owned by my supervising professor. To maintain scientific integrity and retain ownership of my intellectual property, I have to stay independent.
-
Core Mechanism: Autism arises from gene mutations and epigenetic factors that alter regulatory system behavior and constrain neural development and function. Comorbidities arise when stress-responsive BioToggle activation reallocates proteins via epigenetic redox-sensitive BH4 shunt trifurcation, producing predictable comorbidity clusters based on which regulatory system effectors are engaged. The timing and duration of BioToggle activation shape the comorbidity profile, with sustained activation increasing cumulative physiological impact via allostatic overload.
Systems-Level Impact: BioToggles may be situationally triggered by environmental encounters, chronically activated when resolution fails, or genetically locked when mutations impair regulatory reset. Activation generates an allostatic response that is biochemically mediated through BH4-dependent regulation. Under cellular stress conditions GCH1 regulates BH4-dependent trifurcates of three epigenetic, redox-sensitive shunts, producing predictable downstream effects across systems.
The AAAH shunt diverts aromatic amino acids away from dopamine, serotonin, and melatonin synthesis and toward glutamate production via stress-induced transamination. This disrupts excitation-inhibition balance within CSTL circuitry governing movement, habit formation, reward processing, and executive function, thereby altering neural development and behavior.
The NOS shunt reflects redox-regulated, BH4-mediated control of nitric oxide synthase. The BH4 shunt modulates NOS coupling state, permitting regulated NOS uncoupling under stress conditions and shifting nitric oxide production toward reactive oxygen species generation. This redox shift selectively activates downstream epigenetic redox-sensitive protein shunts that function as regulatory system effectors.
The AGO shunt impacts ether lipid cleavage required for endocannabinoid system activity, cellular repair activity, and detoxification.
AGMO is the only enzyme capable of cleaving ether lipid bonds, including plasmalogens, platelet-activating factor, alkylglycerols, and noladin ether. Disruption of noladin ether cleavage to 2-AG alters endocannabinoid signaling. Prolonged AGMO impairment contributes to lysosomal dysfunction, reduced repair capacity, and cumulative system wear.
Epigenetic Redox-Sensitive Protein Shunt Effectors Regulatory Architecture:
The BioToggles are five interdependent regulatory systems that respond to physiological stress through conserved, coordinated activation and context-dependent allostatic resource reallocation to support survival. Activation of the stress response disrupts the BioDials, prioritizing regulatory and survival demands over typical development and typical function in biochemically predictable ways.
The BioDials are four time-regulated protein synthesis mechanisms that maintain the body's steady kinetic flow of protein production across circadian, seasonal, developmental, and lifespan timescales. Preservation of this kinetic flow state is essential for continued physiological function and survival. Prolonged displacement of BioDial-regulated synthesis by stress-responsive BioToggle activity results in allostatic overload with biochemically predictable consequences, the comorbidities.
Individual Variation: Individual differences arise from two mechanisms: experience-driven neural development, which is primarily shaped postnatally by environmental input, and regulatory system activation, in which genetically and epigenetically constrained BioToggles are situationally activated beyond the genetic phenotype. The degree, duration, and timing of this BioToggle activation disrupt BioDial-regulated protein synthesis, reallocating resources toward survival and producing variation in development and function.
Timing Effects: Trait severity and presentation depend on when a gene mutation is functionally relevant, which regulatory system it affects, when BioToggles are activated relative to BioDial-regulated protein synthesis, and how experience shapes neural development and function.
BioToggle activation during sensitive developmental windows more strongly displaces developmental and maintenance processes, while experience-driven neural plasticity during these periods further refines or amplifies functional outcomes.
-
The downstream flow from hypothesis to conclusion is as follows. You can also find how I applied the scientific method here:
Exclusivity Principle
It is implausible for autism traits and comorbid traits to co-occur without a joint root mechanism in each phenotype, arising from the same biochemical mechanism.Gene Mutations
Each phenotype begins with its own mutation pattern.Regulatory System Breach
These mutations impair one or more regulatory systems within the five BioToggles: immune system, metabolism, cellular repair, nervous system, and genetic regulation.Epigenetic Response
The regulatory breach triggers phenotype-specific epigenetic adjustments.Biochemical Pathway Activity
Epigenetic settings reshape pathway behavior. This includes activation of the BH4 Shunt, which reallocates resources across all five BioToggle systems and alters downstream biochemical function according to the specific regulatory disturbance created by the mutation. It also includes the downstream impact of the affected protein within the pathway encoded by that geneBiochemical State
Each phenotype develops a distinct physiological profile shaped by its pathway dynamics.Trait Expression
This physiological state produces both the autism traits and the comorbid traits associated with that phenotype.Individualized Expression
Each individual also encounters environmental experiences that are unique to them as an individual, and thus each individual will also have their own unique profile within their phenotype cluster.
Autism and the Comorbidities Theory
Autism phenotypes can be defined by the allostatic biochemical state produced by their gene mutations. This state explains why autism traits and comorbidities consistently cluster together.Development Into Neurodivergent Biochemistry
BioToggles do not need to be genetically locked to influence traits, they can also be situationally flipped, or chronically stuck. This understanding initiated the development of Neurodivergent Biochemistry. Neurodivergent Biochemistry examines how the body’s survival systems, genetics, and environment work together to shape development, health, and learning. It focuses on how regulatory system activation influences physiological function and developmental outcomes over time.
-
To make the theory teachable and practically useful, I created educational and analytical frameworks for visual and explanatory purposes::
NeuroToggle — neuroplasticity strategies for building, expanding, strengthening, and timing neural connections (used in my daughter’s speech progress).
BioToggles — the five regulatory systems (immune, metabolic, cellular repair, nervous system, genetic regulation).
BioDials — time-based protein synthesis cycles (circadian, circannual, and developmental).
Neurodivergent Biochemistry — a systems-level integration explaining upstream and downstream impacts of allostasis in various contexts considering the variables of the BioToggles becoming situationally flipped, chronically stuck, or genetically locked and how that impacts the timer regulated flow-state of protein synthesis within the BioDials.
Together, these frameworks make the Autism and the Comorbidities Theory and Neurodivergent Biochemistry concepts accessible
-
My work is legally published, copyrighted, and in some cases trademarked:
My website: kimberlyedu.org
My ResearchGate articles on this subject:
Kitzerow, K. (2024a). Autism & the Comorbidities Along the BH4 Pathway. ResearchGate. https://doi.org/10.13140/RG.2.2.23124.37761/1
Kitzerow, K. (2024b). Genomic and Proteomic Regulation in Cellular Homeostasis: From Molecular Mechanisms to Clinical Implications. Research Gate. https://doi.org/DOI: 10.13140/RG.2.2.31853.19682/3
Kitzerow, K. (2025a). Neurodivergent Biochemistry and the Autism and the Comorbidities Theory. Research Gate. https://doi.org/DOI: 10.13140/RG.2.2.30248.89606
The BH4 Pathway as an Allostatic Mechanism in the Pathology of Autism and Systemic Comorbidities. Research Gate. https://doi.org/DOI: 10.13140/RG.2.2.18927.96167
Kitzerow, K. (2025). BioToggle and BioDial categorical delineation: A functional framework for timing-sensitive stress response mechanisms in neurodivergent biochemistry. ResearchGate. https://doi.org/10.13140/RG.2.2.18927.96167
Published Books:
Kitzerow, K. (2023). Discovering autism and the comorbidities along the BH4 pathway. Kindle Direct Publishing.
Kitzerow, K. (2024c). NeuroToggle: Fostering neurodivergent minds through neuroplasticity strategies. Kindle Direct Publishing.
Kitzerow, K. (2025b). NeuroToggle™: A neuroplasticity-based instructional framework. Kindle Direct Publishing. Kitzerow, K. (2024c).
Trademark: 7966373
Videos and visual explanations distributed online to wide audiences (instagram and tiktok)
As an autistic individual and educator with degrees in education and special education with a minor in instruction strategies graduating summa cum laude from UW Superior, I often express complex ideas most effectively through visual formats such as infographics and recorded presentations. These formats are nonetheless publicly available, timestamped, and constitute intellectual property. They establish priority of authorship and fall under the protections of research integrity standards.
Concerning Overlaps With The Four Pillars Of Kitzerow’s Autism And The Comorbidities Theory
What are the 4 Core Pillars of Kitzerow’s Model and How Were They Utilized in 2025 Research?
Genetic and epigenetic mutations activate internal stress-response systems.
Evidence:
A 2025 Japanese study showed diverse autism-associated mutations converge on a common cellular stress response.
Research examining ASD-associated CNVs found shared activation of stress-adaptive signaling pathways across multiple genetic mutations.
Stress-response activation redirects biochemical pathway activity through the BH4 Shunt.
Evidence:
A 2025 Bralian study published a systemic review referring to a theoretical model that redox regulated BH4 and its affected BH4 dependent pathways pathologically link autism and comorbid traits, biochemically linking autism and the comorbidities through the BH4 pathway.
This disruption alters neurotransmitter synthesis and produces excitatory/inhibitory imbalance in the cortico-striatal-thalamic loop, contributing to autism traits.
Evidence:
Stanford research identified reticular thalamus hyperexcitability within CSTL circuitry linked to autism behaviors, and created a successful treatment targeting this mechanism.
Yale research examining glutamate receptors reports findings consistent with E/I imbalance and excitotoxic signaling.
Redox-driven biochemical pathway shifts contribute to systemic comorbidities and phenotypic clustering.
Evidence:
Princeton research showed different categories of genetic mutations alter biochemical pathway activity that clusters autism and comorbid traits into different phenotypic and clinical outcomes.
When evaluating potential overlap with Kitzerow’s framework, it is important to distinguish between three different types of scientific claims related to each of the four pillars of the theory.
A) Mechanisms Of The Four Pillars
This includes research describing the molecular or biochemical mechanisms within the systems involved in the four pillars, such as stress-response signaling, BH4-related biochemical pathways, neurotransmitter synthesis, redox biology, or other related biological processes. These studies explain how individual pathways function at the molecular level.B) Associations Within The Four Pillars
This includes studies reporting correlations between these biological systems and autism. Examples include findings that certain genetic mutations activate stress responses, that BH4 abnormalities appear in autism, that excitatory/inhibitory imbalance occurs in autism circuitry, or that particular biochemical pathways are associated with specific comorbid conditions.C) System-Level Causal Architecture Of The Four Pillars
Kitzerow’s theory proposes a causal architecture linking four steps in sequence: genetic or epigenetic activation of stress-response systems, biochemical pathway redirection through the BH4 shunt, disruption of neurotransmitter balance and CSTL circuitry producing autism traits, and broader biochemical pathway shifts contributing to systemic comorbidities and phenotypic clustering.Mechanistic descriptions (A) and observational associations (B) do not establish the causal architecture described in C. Knowledge of how individual pathways function, or observations that those pathways are altered in autism, does not by itself constitute the system-level model explaining how these four pillars interact to produce autism traits and comorbidity clustering.
Evaluation of overlap therefore concerns whether the causal architecture of any of the four pillars appears in other work. A study does not need to reproduce the entire model for overlap to occur; if the causal structure of one or more pillars is used to explain autism traits or comorbidity clustering, that constitutes overlap with the framework.
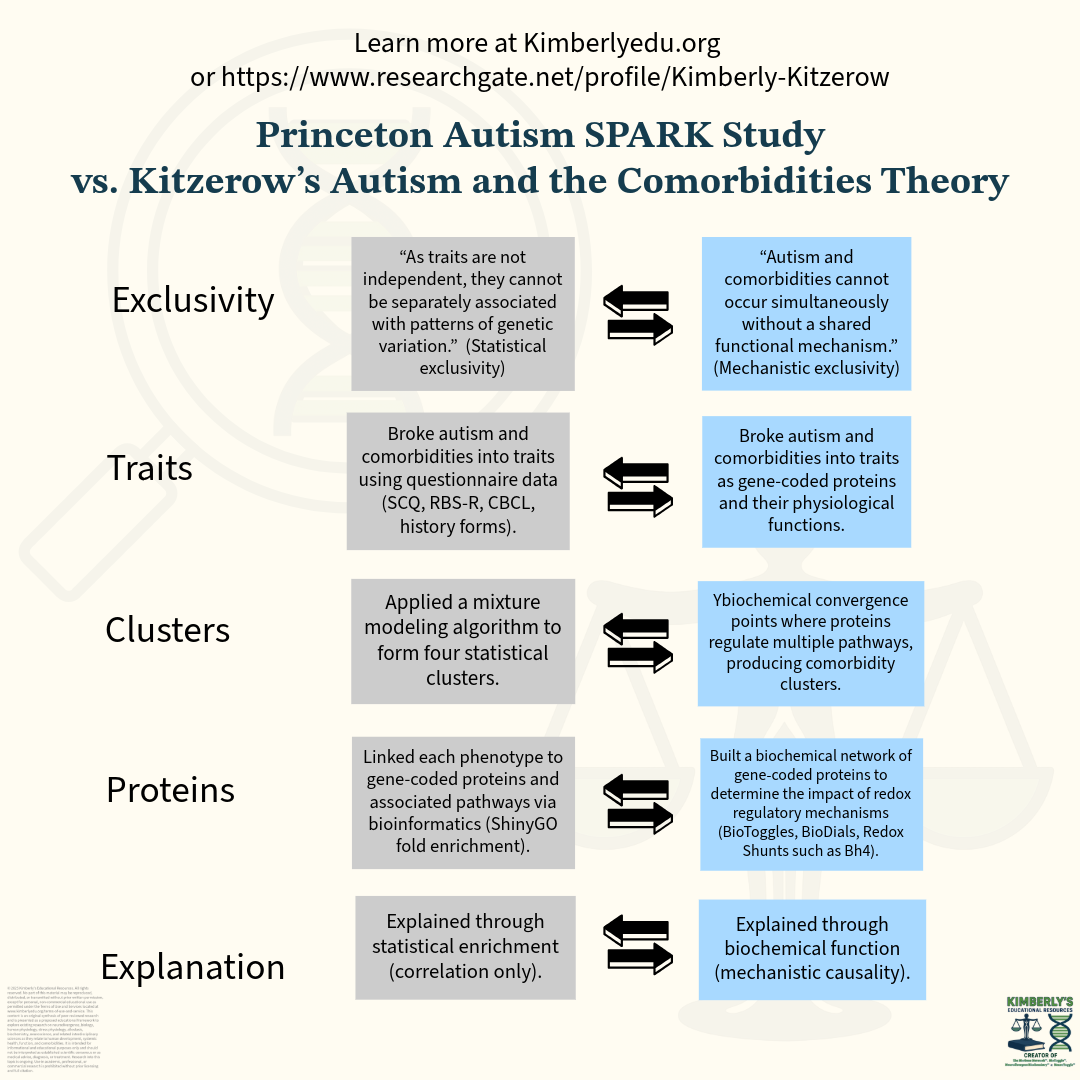
Comparison Between The Princeton Nature Genetics Study “Decomposition Of Phenotypic Heterogeneity In Autism Reveals Underlying Genetic Programs” And Kitzerow’s Autism And The Comorbidities Theory
-
The portion in question is the mechanistic premise that autism trait clusters arise because different gene mutations affect different biological pathways, which in turn produce distinct groupings of autism traits and comorbidities. In Kitzerow’s work, this idea is formalized as the exclusivity principle, which proposes that mutations do not create random mixtures of traits but instead alter biochemical pathway activity in patterned ways that generate predictable phenotypic clusters.
-
Yes. Research has long documented that autism frequently co-occurs with other medical conditions such as gastrointestinal disorders, immune differences, epilepsy, anxiety, and metabolic abnormalities. The presence of comorbidities themselves is not new. What is novel is the proposal that these conditions cluster through shared biochemical pathway mechanisms rather than appearing as unrelated co-occurring conditions.
-
Earlier research typically described comorbidities as conditions that appear alongside autism. Kitzerow’s model instead proposes that the same biological pathway shifts that influence neurodevelopment can also influence systemic physiology. The novel element is the organizing mechanism that explains why specific traits and comorbidities cluster together. In this model, gene mutations alter biological pathway activity in patterned ways, and those pathway differences can produce predictable clustering of autism traits and systemic physiological comorbid traits.
-
Some genetic studies examine whether groups of genes correspond to different biological processes or pathways. The pathway-based model proposes that when genes influence different biological systems, they may produce distinct biological profiles via biochemical pathway activity changes that include both autism traits and particular comorbidities.
-
No. Kitzerow’s theory contains four core pillars, and the presence of BH4 specifically is not required for elements of the framework to appear in other work. The relevant question is whether the causal architecture of any pillar is used. The Princeton study examines whether different categories of genetic mutations alter biological pathway activity and produce distinct autism classes with different clinical and comorbidity patterns. That aligns with Pillar 4 of Kitzerow’s theory, which proposes that genetic variation alters biochemical pathway activity in patterned ways, leading to phenotypic clustering of autism traits and comorbidities. The overlap therefore concerns the pathway-based causal structure explaining trait clustering, not the specific biochemical mechanism involving BH4 described in other parts of the model.
-
They ran their initial GitHub first commit May 24, 2024. This is the mark of the beginning of the project.
Their preprint states their ShinyGO search was ran June 3rd, 2024. In this version they state they tested an identical hypothesis: “Taking the set of impacted genes (genes containing high-impact variants) for each autism class (autism and comorbid clustered phenotypes), we tested the hypothesis that class-specific gene subsets represent distinct pathways and biological processes.”
They submitted their final paper to Nature July 25th, 2024. That is two months from the start of the project to publishing. They ran code for their statistical analysis using existing autism data, for instant results, which you can observe on their GitHub. The statement above about testing this hypothesis was removed in the published version.
You can find the link to my initial hypothesis of the exclusivity principle from 2023 here, and it is also in my book, published September 2023, here. The paper where I investigate the underlying shared biochemical mechanism with different biochemical pathways as they relate to autism and comorbid traits can be found here.
-
Parallel discovery occurs when researchers independently arrive at similar ideas at roughly the same time while investigating the same scientific problem. The concept generally applies to discoveries emerging within a similar timeframe. When a framework was publicly articulated years earlier and later work reflects a similar causal structure, the discussion typically shifts from parallel discovery to questions of plagiarism.
-
In reviewing the Princeton preprint and subsequent Nature publication, there are multiple points of concern:
Hypothesis: In their preprint, it states a hypothesis identical to the Exclusivity Principle: It is implausible for autism traits and comorbid traits to co-occur without a joint root mechanism in each phenotype, arising from the same biochemical mechanism. This mechanistic hypothesis was removed. In the final Nature version, this mechanistic framing was removed, leaving only statistical correlations, although the framework’s structure was retained.
Methodological sequence: I broke down autism and comorbid symptoms into physiological traits, mapped them to protein function, and clustered them via proteins regulating multiple biochemical pathways and the regulatory systems influencing them, thereby generating mechanistic phenotypes. The Princeton version followed the same process but substituted questionnaire-derived traits and statistical clustering, ultimately linking clusters to pathways through correlation-based enrichment.
Post hoc reasoning: A peer reviewer explicitly noted reliance on post hoc reasoning and the absence of functional explanatory logic, underscoring the lack of mechanistic grounding.
Jigsaw Puzzle Methodology: A co-author publicly described their work using the term “jigsaw puzzle,” The Autism and the Comorbidities Theory was created using the the Jigsaw Puzzle Research Methodology, as described in a prior publication. This is due to the nature of piecing together a biochemical network out of raw protein data from Uniprot, and comparing autism biomarkers to find points of dysregulation liek a puzzle. The Princeton study similarly decomposes traits, clusters them, and retrospectively aligns them with biochemical pathways, but does so without acknowledgment of that methodology or its mechanistic structure.
This section compares the 2025 Princeton Nature Genetics study, “Decomposition of Phenotypic Heterogeneity in Autism Reveals Underlying Genetic Programs” (DOI: https://doi.org/10.1038/s41588-025-02224-z), with Kitzerow’s 2023 Autism and the Comorbidities theoretical model and Exclusivity Principle hypothesis.
The Princeton study tests the hypothesis that categories of gene mutations alter biochemical pathway activity and cluster autism and comorbid traits into distinct phenotypic groups. Kitzerow’s Exclusivity Principle hypothesissimilarly proposes that genetic variation shifts biochemical pathway activity in patterned ways, producing predictable clustering of autism traits and systemic comorbidities.
Learn more about Kitzerow’s Autism and the Comorbidities theory here.
Comparison Between The Stanford Study “Reticular Thalamic Hyperexcitability Drives Autism Spectrum Disorder Behaviors In The Cntnap2 Model Of Autism” And Kitzerow’s Autism And The Comorbidities Theory
This section compares the Stanford study “Reticular thalamic hyperexcitability drives autism spectrum disorder behaviors in the Cntnap2 model of autism” (DOI: http://doi.org/10.1126/sciadv.adw4682) with Kitzerow’s Autism and the Comorbidities theoretical model.
The Stanford study examines whether reticular thalamic hyperexcitability drives autism behaviors and reports that reducing this circuit-level hyperactivity improved autism-like traits in a mouse model. Kitzerow’s framework similarly places excitatory/inhibitory dysregulation within neural circuits as the downstream mechanism producing autism traits. This section also notes the 2023 outreach from Stanford researchers requesting information about Kitzerow’s framework and the subsequent 2025 development of a treatment targeting that same circuit-level mechanism.
Learn more about Kitzerow’s Autism and the Comorbidities theory here.
-
The concept that excitatory/inhibitory imbalance functions as a downstream effect within a broader regulatory stress cascade. In Kitzerow’s model, genetic or epigenetic activation of stress-response systems leads to biochemical pathway shifts, which ultimately disrupt excitatory/inhibitory balance within specific neural circuits, producing autism traits. The Stanford study targeted excitatory/inhibitory imbalance within a defined cortico-striatal-thalamic circuit segment. The alignment lies in the positioning of E/I dysregulation as a key downstream mechanism contributing to autism behaviors.
Kitzerow petitioned Stanford’s Neurodiversity Project in January of 2024 for help, and they recently published a paper utilizing the downstream mechanism of upregulated transamination pathways from the BH4 Shunt dysregulating E/I balance in a piece of the CSTL to create an effective “cure” for autism’s classic core symptoms.
-
The Stanford study targeted hyperexcitability in the reticular thalamic nucleus (RT), a brain region within the cortico striatal thalamic loop that regulates thalamocortical signaling. In the autism mouse model (Cntnap2 knockout mice), neurons in this region were firing too easily and too frequently. The researchers reduced this excessive activity using a T-type calcium channel blocker called Z944, which decreases burst firing in those neurons. They also used a research tool called chemogenetic inhibition (hM4Di activated by C21) to suppress the same neurons. When RT excitability was reduced, the mice showed improvements in autism-related behaviors such as repetitive behaviors, hyperactivity, and social deficits.
-
Think of brain signals like traffic lights.
Green lights tell signals to go.
Red lights tell signals to stop.In the mice, there were too many green lights and not enough red lights, so brain signals were firing too much. The drug helped restore balance by reducing the extra green-light activity, calming the circuit.
It targets the final step in the model. In Kitzerow’s framework, upstream biological stress changes eventually cause too many green signals (excitation) and not enough red signals (inhibition) in certain brain circuits. That imbalance produces autism traits. The Stanford treatment works by correcting that final signal imbalance in the circuit.
-
In 2023 someone from Stanford’s Neurodiversity Project reached out to Kitzerow requesting information about her model. She obliged.
Kitzerow petitioned Stanford’s Neurodiversity Project in January of 2024 for help advancing this model.
In 2025 they recently published a paper utilizing the downstream mechanism of upregulated transamination pathways from the BH4 Shunt dysregulating E/I balance in a piece of the CSTL to create an effective “cure” for scription
-
You can find Kitzerow’s breakdown of the BH4 Shunt induced upregulated transamination pathways that dysregulate E/I balance in the CSTL, which is the brain circuitry for movement, habit formation, and reward, leading to classic autism traits published August 2024 here. You can watch the videos of Kitzerow explaining this E/I dysregulation induced autism trait mechanism for the first time June 2023 here and here.
The link between E/I imbalance in the CSTL and autism has been known for a while, I am the first to link it into a cohesive framework centered around what is causing this E/I imbalance.
You can find Stanford’s 2025 version here.
-
No. Because the intervention acts only at the circuit level, it addresses the behavioral output of the cascade. In Kitzerow’s framework, systemic comorbidities arise from simultaneous biochemical pathway shifts driven by redox changes and the cumulative wear and tear of allostatic overload from chronic regulatory system activation. Correcting the final neural circuit imbalance would not be expected to reverse those upstream systemic processes.

Comparison Between The Harvard / Northeastern Study “It’s Not The Thought That Counts: Allostasis At The Core Of Brain Function” And Kitzerow’s Autism And The Comorbidities Theory
-
The BioToggles are five interdependent regulatory systems that respond to physiological stress through conserved, coordinated activation and context-dependent allostatic resource reallocation to support survival.
Their paper states: “In allostasis, many interacting regulatory systems collectively contribute to the survival of the organism.”
The concept of categorical regulatory systems operating together to maintain biological stability. In Kitzerow’s framework, the body’s regulation is organized into distinct but interacting systems such as the nervous system, immune system, metabolism, cellular repair, and genetic regulation. These systems function interdependently and respond to stress together. The Northeastern/Harvard paper similarly describes allostasis as the coordinated activity of multiple interacting regulatory systems and identifies the nervous system as one component within that broader regulatory structure.
The overlap lies in the multi-system regulatory architecture, where regulation is framed as the interaction of distinct biological systems rather than a single unified network.
-
The 2025 paper reframes allostasis as the coordinated activity of multiple interacting regulatory systems. In this model, the nervous system is described as one regulatory component that operates interdependently with other categorical regulatory systems.
Kitzerow categorically delineated regulatory system domains based on patterns of protein induction under redox conditions while constructing her biochemical network. This categorical structure represents a different way of organizing allostasis conceptually, framing regulation as the interaction of multiple system domains rather than a single integrated process. In her framework, these domains are referred to as BioToggles, which include the immune system, metabolism, cellular repair, nervous system, and genetic regulation.
-
Earlier literature generally described allostasis as a single integrated regulatory process distributed across interoceptive and visceromotor networks. The emphasis was on a unified system with interconnected feedback loops maintaining stability, rather than on distinct categorical regulatory systems interacting with one another.
-
Supporting Materials:
Exhibit A Harvard: New multi-system regulatory framing
Quote: “In allostasis, many interacting regulatory systems collectively contribute to the survival of the organism.”
Explanation: First appearance of plural regulatory systems in Harvard’s work. Establishes a categorical multi-system architecture aligned with the BioToggles model.
Exhibit B Harvard: Prior unified allostatic system (October 2024)
Quote: “These results reinforce the existing evidence for a whole-brain system that supports the modeling and regulation of the body’s internal milieu.”
Explanation: Reinforces their historical single-system framing before the introduction of plural regulatory systems in 2025.
Quote: “Large swaths of brain tissue are centrally involved in allostasis, with the rest serving as association regions for the predictive, efficient regulation of the body.”
Explanation: Separates core allostatic regions from distinct body-regulatory regions, creating categorical regulatory functions rather than a unified system. This represents an early move toward multiple regulatory systems.
-
You can find the BioToggles framework here. It was first developed into the allosatic toggles structure here. The written explanation can be found in this paper. Trademark was filed April 2025, and the serial number is 99148316.
You can find their paper that outlines this mechanism here.
Their prior paper that doesn’t include the term regulatory system, or the novel concept of interdependent regulatory systems here.
This section compares the Harvard / Northeastern study “It’s Not the Thought That Counts: Allostasis at the Core of Brain Function” (DOI: http://doi.org/10.1016/j.neuron.2025.09.028) with Kitzerow’s Autism and the Comorbidities theoretical model.
Prior literature generally described the regulatory system involved in allostasis as a single integrated unit, with feedback loops operating within interoceptive and visceromotor networks to maintain physiological stability. The 2025 Harvard / Northeastern paper instead describes allostasis as a multi-regulatory system mechanism that maintains survival. Kitzerow’s framework similarly organizes biological regulation into distinct interacting regulatory system domains, referred to as BioToggles, including the immune system, metabolism, cellular repair, nervous system, and genetic regulation. This section compares the multi-regulatory system architecture described in the Harvard / Northeastern study with the categorical regulatory system structure articulated in Kitzerow’s model.
Learn more about Kitzerow’s Autism and the Comorbidities theory here.

UCSD: Naviaux’s New 3-Hit Model vs Kitzerow’s Autism and the Comorbidities Theory
-
The concept of a three-category stress taxonomy initiating a structured biological cascade that progresses through the same downstream nodes. In Kitzerow’s framework, three stress sources (genetic, chronic, and situational) initiate a sequenced cascade that moves through mitochondrial and redox shifts, excitatory/inhibitory dysregulation, autism traits with predictable comorbidities, developmental timing, and neuroplastic adaptation. Naviaux’s 2025 Three-Hit model similarly begins with three stress categories and proceeds through mitochondrial or metabolic shifts, excitatory/inhibitory dysregulation, autism and comorbidities, developmental timing, and neuroplasticity. The alignment therefore concerns not only the presence of stress biology but the same ordered cascade architecture linking these downstream mechanisms.
-
From approximately 2013 through 2024, Naviaux’s framework centered on the Cell Danger Response (CDR) and followed a relatively consistent structure: stress activation leading to mitochondrial or metabolic shifts that contribute to chronic multi-system disease. The framework did not formally include a defined three-category stress taxonomy, a sequenced excitatory/inhibitory dysregulation node, a structured autism plus comorbidity simultaneity model, a developmental timing node positioned after autism emergence, or neuroplasticity as a terminal adaptive mechanism.
-
Kitzerow’s published framework presents an ordered cascade beginning with three stress-state categories (genetic, chronic, situational). These lead to a BH4 pathway redistribution, producing redox and mitochondrial shifts alongside excitatory/inhibitory dysregulation. This cascade produces autism traits and predictable comorbidity clusters, followed by a developmental timing phase and ultimately neuroplastic adaptation through NeuroToggle®.
-
Conceptual Reframing: Naviaux has shifted from describing CDR as a unified metabolic response to describing autism and its comorbidities as a staged biological progression. This new structure reflects the BioToggle sequence of situationally flipped, chronically stuck when resolution fails, and genetically locked. None of this appears in his earlier work.
Adoption of the Autism and Comorbidities Framework: He now presents autism and its comorbidities as a single mechanistically connected pathological framework. His statement that ASD is associated with “an increased risk of several chronic medical conditions that co-occur with ASD” adopts the exact framing, and verbiage, that my Autism and the Comorbidities Theory introduced. He had never previously offered a mechanism that links autism traits and comorbidities into a single framework.
Mechanistic Adoption: The updated paper introduces BH4 dependent stress regulation of the excitatory to inhibitory balance. Naviaux states that prolonged activation “prevents the normal excitatory to inhibitory reversal in extracellular ATP and GABA signaling networks” and then links this failure to mitochondrial dysfunction and activation of microglia and astroglia. This mechanism is a core component of my BH4 Shunt model.
Use of PKU While Avoiding BH4: The update uses PKU as the model’s key developmental analogy and states that “the three hit model described above for ASD also applies to the current paradigm for the diagnosis and treatment of phenylketonuria (PKU)”. Earlier in the paper he explains that the enzyme responsible for phenylalanine metabolism requires the cofactor tetrahydrobiopterin. He writes that PAH converts phenylalanine to tyrosine using “iron, molecular oxygen, and the cofactor tetrahydrobiopterin”.
PKU is a BH4 dependent disorder. Using PKU to illustrate early biochemical disruption while avoiding any mention of BH4 within the autism mechanism is scientifically inconsistent unless the upstream mechanism is being intentionally avoided. The BH4 Shunt is the central biochemical mechanism I am known for, and PKU is one of its clearest clinical demonstrations.
Excitatory to Inhibitory Imbalance Integration: The updated paper positions failure of the excitatory to inhibitory reversal as a core downstream consequence of prolonged CDR activation. The relevant quote is that prolonged activation “prevents the normal excitatory to inhibitory reversal in extracellular ATP and GABA signaling networks”. He does not specify what this mechanism is. In my framework the BH4 Shunt upregulates transamination pathways that dysregulate this balance through unregulated glutamate activity and synthesis, dysregulatin the cortico-striatal-thalamic-loop that is responsible for movement, habit formation, and reward, thus resulting in core autism traits. This is one of the defining downstream effects of BH4 dysregulation in my published work. This mechanistic connection does not appear in earlier CDR publications.
-
The concern does not center on individual shared concepts such as stress or mitochondria. Instead, it focuses on ordered scaffolding across multiple nodes. When several elements appear in the same directional progression within a framework that previously followed a different structure, the alignment becomes architectural rather than incidental.
-
You can find the BioToggles framework here. It was first developed into the allosatic toggles structure here. The written explanation can be found in this paper. Trademark was filed April 2025, and the serial number is 99148316.
You can find Kitzerow’s breakdown of the BH4 Shunt induced upregulated transamination pathways that dysregulate E/I balance in the CSTL, which is the brain circuitry for movement, habit formation, and reward, leading to classic autism traits published August 2024 here. You can watch the videos of Kitzerow explaining this link for the first time June 2023 here and here. She also had a viral Instagram video August 30, 2024 breaking this down that you can watch here.
You can find Kitzerow’s first mention of time regulated impact of stress on development here. She developed it over timeturning it into the BioDials framework as described here.
You can find the link to Naviaux’s 2025 3-Hit Model here, and his prior version of CDR here and here.
This section compares the study “A 3-Hit Metabolic Signaling Model for the Core Symptoms of Autism Spectrum Disorder” (DOI: https://doi.org/10.1016/j.mito.2025.102096) with Kitzerow’s Autism and the Comorbidities theoretical model.
Naviaux’s 2025 paper presents an updated version of the Cell Danger Response (CDR) model, which had remained structurally consistent since its introduction in 2014. The updated framework introduces a three-hit stress taxonomy and an expanded downstream signaling sequence involving mitochondrial signaling, neural regulation, developmental timing, and neuroplastic adaptation.
Kitzerow’s 2023 Autism and the Comorbidities Theoretical Model similarly begins with three initiating stress states (genetic, chronic, and situational) and describes a structured biochemical cascade involving BH4 pathway redistribution, mitochondrial and redox shifts, excitatory/inhibitory dysregulation, autism traits with predictable comorbidities, developmental timing, and neuroplastic adaptation. This section compares the cascade architecture introduced in Naviaux’s 2025 update with the ordered biochemical cascade previously articulated in Kitzerow’s model.
Learn more about Kitzerow’s Autism and the Comorbidities theory here.

Comparison Between The Systematic Review “Tetrahydrobiopterin And Autism Spectrum Disorder: A Systematic Review Of A Promising Therapeutic Pathway” And Kitzerow’s Autism And The Comorbidities Theory
-
“Neurobiological hallmarks of ASD include abnormal synaptic connectivity, imbalances in excitatory–inhibitory signaling, and alterations in neurotransmitter systems such as serotonin, dopamine, glutamate, and gamma aminobutiric acid (GABA). Immune dysfunction, chronic inflammation, oxidative stress, and metabolic abnormalities, including mitochondrial dysfunction, have been described as critical factors. Considering that many of these molecular and signaling pathways may be dependent on the availability of 6R-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4), alterations in its metabolism have been implicated in the pathogenesis of ASD.”
and provides no citation for the source.
Kitzerow’s Autism and the Comorbidities Theory (2023) is the first to mechanistically delineate how and why the BH4 Shunt physiologically manifests into autism and comorbid traits.
-
Use of this hypothesis as prior knowledge without citation
In their introduction, the authors listed this hypothesis as though it were already established knowledge, without mentioning where it came from. MDPI was contacted and Latini claims the hypothesis was a conclusion to this study. How can it be both hypothesis and a novel conclusion drawn from the results of the systematic review?Methodological and registration issues
The methods section acknowledges the review was not registered during the planning stage, which increases the risk of duplication. Their search strategy was limited to BH4, nitric oxide, and ASD, excluding other BH4-dependent pathways required to construct the complete Autism and the Comorbidities framework. Despite this, the paper presents the completed theory as though it had been their intent all along. There is also no public record of the work before their December 31, 2024 submission to MDPI, and it relied on a universal grant with no detailed proposal.Prior Work on BH4 Published Prior to Kitzerow’s Theory Lists BH4 in the “Non-BH4-Linked Genetic Deficiencies of BH4 Metabolism” Section: Latini’s prior paper on BH4 and its role as a cofactor with no mention of a causal role in autism and the comorbidities, only a mention of autism in a section titled “Non-BH4-Linked Genetic Deficiencies of BH4 Metabolism” here. It was submitted to publishing March 2023, and published May 2023. Had she developed this theory prior, it would have been listed differently in that paper.
-
Latini previously discussed BH4 in autism within a section titled “Non-BH4-Linked Genetic Deficiencies of BH4 Metabolism” in the paper Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor, which the journal received on March 28, 2023. In that work, BH4 was primarily described in its traditional biochemical role as a cofactor in metabolic pathways and was cons
Latini’s earlier view described BH4 primarily as a metabolic cofactor influencing neurotransmitter synthesis, particularly monoaminergic pathways, in autism. Reduced BH4 levels in some children with ASD were interpreted as contributing to symptoms through effects on dopamine and serotonin production, potentially driven by inflammation, oxidative stress, immune activation, folate metabolism, or rare metabolic gene variants. This positioned BH4 near the intersection of several biological processes, but it was discussed mainly as a metabolic contributor or therapeutic target, rather than as a redox-sensitive regulatory mechanism coordinating multiple BH4-dependent pathways across autism and its systemic comorbidities..
-
The 2025 review expands BH4’s role beyond a metabolic cofactor affecting monoamine neurotransmitter synthesis. Instead, it frames BH4 as a redox-sensitive regulatory mechanism influencing multiple biological pathways involved in autism, including nitric-oxide signaling, immune activation, oxidative stress, and metabolic function.
In this updated framing, autism traits and associated comorbid conditions are discussed as emerging from BH4-dependent pathway disruptions that occur under inflammatory and oxidative stress conditions, linking neurotransmitter imbalance, immune dysregulation, metabolic abnormalities, and mitochondrial dysfunction within a shared biochemical context.
-
A key concern relates to the structure and evidentiary basis of the framework presented in the paper. Systematic reviews typically contain two distinct citation domains: the introductory literature review, which provides scientific context and may include studies published up to the time of submission, and the systematic review analysis, which is limited to the studies captured through the defined search strategy.
In this case, the paper presents a broad conceptual model describing BH4 as a redox-regulated mechanism pathologically linking autism and comorbid traits, including effects on neurotransmitter systems, nitric oxide signaling, immune activation, oxidative stress, and metabolic dysfunction. However, the systematic review itself searched only for studies involving BH4, neopterin, nitric oxide, and related metabolites in autism literature through December 2021.
This raises questions about how the broader pathway-level model was constructed. A redox-regulated framework would normally require systematic evaluation of oxidative stress markers and redox biomarkers, yet those markers were not the primary search terms used in the review methodology. Similarly, the model discusses multiple BH4-dependent biological pathways, but the search strategy did not systematically examine the full set of biomarkers associated with those pathways.
The paper also states that the systematic review was not preregistered during the planning stage, which increases the possibility of duplication or overlap with previously published conceptual models.
As a result, the concern being raised is not whether dysregulated BH4 levels have been associated with autism, which earlier literature already suggests, but whether the comprehensive redox-regulated BH4 pathway framework pathologically linking autism and systemic comorbidities presented in the paper extends beyond what can be derived from the systematic review methodology described.
-
-
You can find the link to the first video version of Kitzerow’s Autism and the Comorbidities here, and the first paper here.
You can find Latini’s prior paper on BH4 and its role as a cofactor with no mention of a causal role in autism and the comorbidities, only a mention of autism in a section titled “Non-BH4-Linked Genetic Deficiencies of BH4 Metabolism” here. It was submitted to publishing March 2023, and published May 2023. Had she developed this theory prior, it would have been listed differently in that paper.
Latini’s updated 2025 paper with the BH4 Shunt Autism and the Comorbidities Hypothesis can be found here.
-
The overlap occurs in the pathway-level framing of BH4 as a redox-regulated mechanism that pathologically links autism traits with systemic comorbidities.
Kitzerow’s model describes a redox-regulated BH4 Shunt in which stress-driven changes in BH4 availability simultaneously alter multiple BH4-dependent pathways, including neurotransmitter synthesis, nitric oxide signaling, immune activation, and metabolic regulation. In this framework, those biochemical pathway shifts produce autism traits and associated comorbid conditions at the same time.
The 2025 paper similarly describes BH4 as a redox-sensitive mechanism connecting neurotransmitter systems, nitric oxide signaling, immune dysfunction, oxidative stress, and metabolic abnormalities in autism, placing these processes within a shared biochemical context.
The conceptual alignment therefore occurs in the redox-regulated BH4 pathway architecture linking autism and systemic comorbidities through coordinated biochemical pathway shifts, rather than viewing BH4 solely as an isolated metabolic cofactor or supplementation target.
This section compares the paper “Tetrahydrobiopterin and Autism Spectrum Disorder: A Systematic Review of a Promising Therapeutic Pathway” (DOI: https://doi.org/10.3390/brainsci15020151) with Kitzerow’s Autism and the Comorbidities theoretical model.
The 2025 systematic review examines literature on BH4 metabolism in autism and presents a framework in which the BH4 pathway is pathologically implicated in autism and associated comorbid conditions. The paper discusses BH4 as a redox-sensitive mechanism influencing multiple biological systems, including neurotransmitter regulation, nitric oxide signaling, immune activation, oxidative stress, and metabolic dysfunction, and describes how disruptions in this pathway may contribute to both autism traits and the clustering of comorbidities. This section evaluates how that BH4-centered pathological pathway framework linking autism and comorbid traits compares with the redox-regulated BH4 Shunt and resulting biochemical cascade articulated in Kitzerow’s model.
Learn more about Kitzerow’s Autism and the Comorbidities theory here.

Comparison Between Nitric Oxide-Mediated S-Nitrosylation Of TSC2 Drives mTOR Dysregulation Across Shank3 And Cntnap2 Models Of Autism Spectrum Disorder And Kitzerow’s Autism And The Comorbidities Theory
-
The concept utilized is the epigenetic redox-sensitive protein shunt, specifically the mTOR shunt. In Kitzerow’s model, redox-dependent chemical modifications alter the stability or activity of regulatory proteins that control major signaling pathways. This paper demonstrates that mechanism by showing that nitric oxide chemically modifies the regulatory protein TSC2 through S-nitrosylation. Once modified, TSC2 becomes unstable and is degraded, removing one of the primary inhibitory controls on the mTOR pathway and allowing mTOR signaling to become overactive. Because TSC2 is a central regulator of mTOR activity, the redox-driven modification of this protein shifts the signaling state of the mTOR pathway, reflecting the mechanism described in the mTOR epigenetic redox-sensitive protein shunt.
-
The study identified a nitric oxide–driven redox mechanism in which the protein TSC2 undergoes S-nitrosylation. This chemical modification destabilizes the protein and leads to its degradation. Because TSC2 normally functions as a brake on mTOR signaling, its loss removes inhibitory control and allows the mTOR pathway to become overactive.
-
No. Kitzerow’s model does not propose simple BH4 depletion. It proposes a BH4 shunt, where BH4-dependent enzymatic resources are redistributed under redox stress conditions. In this state, the coupling efficiency of nitric oxide synthase (NOS) can change. Some NOS activity may still produce nitric oxide, while uncoupled NOS can generate superoxide. These molecules can also interact to form reactive oxygen species such as peroxynitrite that drive nitrosative modifications such as S-nitrosylation. In this framework, the shift is not random but need-driven, meaning biochemical resources are redirected in response to cellular stress conditions.
-
Within Kitzerow’s framework, the BH4 shunt alters NOS behavior under redox stress conditions. Changes in NOS coupling can increase both nitric oxide signaling and the production of reactive oxygen species. These redox conditions promote cysteine modifications such as S-nitrosylation, the same chemical modification shown in the study to destabilize TSC2 and activate mTOR signaling.
-
The mTOR pathway regulates synaptic protein synthesis, neuronal growth, and synaptic pruning during development. When redox-driven protein modifications destabilize regulators like TSC2, mTOR signaling can become overactive, altering neural circuit formation and development.
-
This section compares the 2026 study “Nitric Oxide-Mediated S-Nitrosylation Of TSC2 Drives mTOR Dysregulation Across Shank3 And Cntnap2 Models Of Autism Spectrum Disorder” (DOI: https://doi.org/10.1038/s41380-026-03514-6) with Kitzerow’s Autism and the Comorbidities theoretical model.
The study demonstrates that nitric oxide–driven S-nitrosylation chemically modifies the regulatory protein TSC2, destabilizing the protein and removing inhibitory control over the mTOR signaling pathway. In Kitzerow’s model, Epigenetic Redox-Sensitive Protein Shunts describe how redox-dependent chemical modifications to regulatory proteins shift the activity state of major biological pathways. This section evaluates how the NO → TSC2 → mTOR mechanism described in the study compares with the mTOR Epigenetic Redox-Sensitive Protein Shunt articulated in Kitzerow’s framework.
Learn more about Kitzerow’s Autism and the Comorbidities theory here.